本帖最后由 costa_na 于 2013-12-24 17:28 编辑
MET inhibition in lung cancer
MET抑制剂在肺癌中的应用
注:这篇综述太长了,选取了一部分有价值的翻译给大家看看
Met discovery and mechanism of action
Met的发现和作用机制
Met is a heterodimer receptor tyrosine kinase composed of a α-chain and a β-chain, linked by a disulphide bond.
Met是一种二聚的受体酪氨酸激酶,由一个α链和一个β链通过二硫键链接而成

从这张图可以看到α链和β链,磷酸根的转移发生于Tyr 1234和Tyr 1235,而Tyr 1349和Tyr 1356能插入到多底物的停泊位点,从而通过募集SH2链接分子启动下游信号转导
Met was originally isolated as the product of a human oncogene, trp-met, in tumor cells treated with a chemical carcinogen. Met gene encodes a 170-kD protein (p170met) that has constitutive and ligand-independent tyrosin kinase activity. Met has pivotal functions in embryogenesis and organogenesis of placenta, liver, kidney, neurons and muscles (22-25).
Met最开始作为人类原癌基因trp-met的产物,在由化学致癌物诱导的癌细胞中被分离出来。Met基因编码一个170kD的蛋白质,该蛋白质具有结构化并且独立于配体的磷酸化激酶活性。Met在胚胎形成和如胎盘、肝脏、肾脏、神经和肌肉等器官生成等功能上具有关键性作用。
Moreover, in vivo, Met receptor activation determines a phenomenon called “invasive growth”, which includes cell proliferation, scattering, survival, motility and invasion, epithelial-mesenchymal transition and branched morphogenesis (26,27).
而且,Met受体激活在体内表现出一种称为“侵袭性生长”的现象,主要包括细胞的增值、散播、生存、移动和侵袭、上皮间质转化(EMT)和分支形态生成。
The natural ligand for this receptor is the HGF, produced by stromal and mesenchymal cells, that acts primarily on Met-expressing epithelial cells in an endocrine and/or paracrine fashion (24,28). HGF-induced Met tyrosine kinase activation is regulated by paracrine ligand delivery, ligand activation at the target cell surface and ligand-activated receptor internalization and degradation (29). Going more into details, when HGF binds to the Met receptor, Met major autophosphorylation sites (located within the tyrosine kinase domain) are phosphorylated, with subsequent intrinsic catalytic activation of multiple signaling cascades involved in cell proliferation, survival, angiogenesis, morphogenesis, cell scattering, motility, migration and invasion. An activated docking site in the kinase domain further recruits intracellular adaptor molecules through the SH2 domains and other recognition motifs, such as GAB1 (a key coordinator of the cellular responses to Met). Downstream signaling of the GRB2-mitogen-activated protein kinase (MAPK) cascade, PI3K-mTOR pathway, and STAT pathway are eventually activated, mediating various cellular functions (27,30,31). Finally, in order to activate the receptor, proteolytic cleavage of proHGF is necessary (25).
该受体的自然配体是肝细胞生长因子(HGF),由基质和间质细胞产生,主要以内分泌和旁分泌的方式作用于表达Met的上皮细胞。HGF诱导的Met酪氨酸激酶的激活通过三种方式进行调节:旁分泌配体输送、在靶细胞表面的配体激活和配体激活后受体的内在化和降解。更详细的描述是,当HGF绑定到Met受体之后,Met主要自磷酸化位点(位于其酪氨酸激酶域中)磷酸化,导致一系列涉及细胞增值、生存、血管生成、形态发生、细胞散播、移动、迁移和侵袭的下游级联通路的催化激活。在激酶域的一个激活的停泊位点将会通过SH2域募集胞内的链接分子和其他识别基序如GAB1(作为细胞对Met响应的一个关键的调节器)。最终,下游信号如GRB2-MAPK级联、PI3K-mTor通路和STAT通路将被激活,介导多种细胞功能。最后,要激活受体,proHGF还需要经过蛋白酶水解切割。

Met和其下游信号通路的一个总体描述图
HGF is mainly produced by stromal tissue like liver and bone marrow, and is expressed in a multitude of mesenchymal-derived cells. Being Met expression detected in the epithelium of most tissues, this indicates that HGF-Met signal transduction pathway contributes to mesenchymal-epithelial interactions (24,32-34).
HGF主要由基质组织产生如肝脏和骨髓,能够在大量的间质来源的细胞中表达。因为在绝大多数组织的上皮细胞中检测到了Met的表达,这表面HGF-Met信号转导促使了上皮间质转化过程。
Met downregulation occurs through rapid internalization of Met itself and subsequent degradation by the lysosome: this process is regulated by ligand-dependent ubiquitination of Met, a process also modulated by specific tyrosine phosphatases and recently identified as proteins decorin and LRIG1 (35,36).
Met的下调发生于Met自身的快速内在化以及随后被溶酶体降解:配体依赖的Met泛素化调节该过程,而泛素化又受到特定的酪氨酸磷酸化酶的控制,最近研究识别出该酶为胶饰蛋白多糖和LRIG1。
Met can be altered through receptor overexpression, genomic amplification, mutations or alternative splicing. These alterations lead to signaling deregulation that can be mediated through ligand (HGF)-independent receptor activation or through its ligand (HGF)-dependent activation via autocrine (intratumoral HGF), paracrine (mesenchymal or microenvironmental HGF), or endocrine (circulatory HGF) loop signaling cascades (29).
Met可以通过受体的过表达、基因组的扩增、突变和选择性剪切被改变。这些变化导致了信号的失调,该失调是受两种作用的介导:不依赖于配体的受体激活或者依赖于配体的受体激活,配体依赖的激活主要通过自分泌(瘤内的HGF)、旁分泌(间质或者微环境中的HGF)和内分泌(血液循环中的HGF)方式循环进行的信号级联反应。
HGF and Met are highly expressed in various stem and progenitors cells, but are only expressed as low levels in their mature cells (25). In preclinical animal models, whereas the overexpression of Met and/or HGF has been shown to stimulate tumorigenesis and metastasis, downregulation of Met or HGF expression resulted in increased apoptosis and decreased tumor growth and blood vessel density (37-40). Moreover, Met interacts synergistically with VEGF to promote angiogenesis, cell proliferation and invasion (41). This occurs through the transcriptional upregulation of the hypoxia inducible factor-1α and amplified HGF signaling, that resulted in both induction of invasion and increased expression of VEGF (41).
HGF和Met在多种干细胞和前体细胞中存在高表达,但在成熟细胞中表达水平很低。在亚临床动物模型中,Met和/或HGF的过表达表现出对肿瘤生长和转移的刺激作用。对Met和HGF的下调导致了细胞凋亡的增加以及肿瘤成长速度和血管密度的降低。而且,Met能协调地作用于VEGF从而促进了血管生成、细胞增值和侵袭。通过对缺氧诱导因子(HIF-1α)转录下调和HGF的扩增,导致了侵袭的发生和VEGF表达的增加。
Met pathway is also one of the key players in the development of acquired resistance to VEGF pathway inhibitors: the inhibition of Met expression prevented hypoxia-induced invasion growth (42,43).
Met通路也在对VEGF抑制剂的耐药的产生过程中扮演了关键角色:对Met表达的抑制阻止了缺氧诱导的侵袭性生长。
The increased Met expression described in case of response to ionizing radiation through the ATM-NFκB signaling pathway, could lead to radioresistance and cancer invasion (44).
Met能够通过ATM-NFkB信号通路增加表达以阻止对电离辐射的响应,这可以导致对放疗的抵抗和肿瘤的侵袭
Met pathway and cross-talks
Met通路和与其他信号通路的交互作用
The cross-talk of Met with various signaling pathways is described in literature and that one between Met and EGFR/HER family receptors is particularly important in lung cancer (45-49).
Met与其他多种信号通路的交互作用已经在文献中得到了阐述,在肺癌中,Met和EGFR/HER家族的交互表现得特别重要。
Met and EGF family receptors are often described co-expressed in tumors and transactivation of Met depends on elevated expression of EGFR in many human tumors (46,50,51). Conversely, HGF stimulation promotes transactivation of EGFR in multiple cell lines, including NSCLC (49).
在许多人类肿瘤中都发现了Met和EGF族受体的协同表达以及依赖于EGFR表达水平提升的Met转录活化。与此对应的,HGF的刺激也能在包括NSCLC在内的多种细胞系中促进EGFR的转录活化。
Cooperation between Met and EGFR occurs also indirectly: when Met activates Src, this lead to EGFR phosphorylation and the creation of docking sites for EGFR interactors involved in downstream signaling (52).
Met和EGFR的协同作用总是间接的:Met对Src的激活会导致EGFR的磷酸化以及涉及下游信号的EGFR交互子的停泊位点的建立。
Moreover, through receptor cross-talk, Met exerts a key role in the development of resistance to EGFR family inhibitors. One example is the stimulation of HER-3 phosphorylation and signaling to Akt (a key signaling molecule required for cell survival and proliferation) when Met is amplified and overexpressed (53,54). Inhibition of Met in EGFR inhibitors resistant cells, either in vitro or in vivo, promotes apoptosis, tumor growth reduction and signifcant necrosis (49,53).
而且,通过受体的交互作用,Met在对EGFR家族抑制剂耐药性发展的过程中也表现出了关键的作用。一个例子就是Met的扩展会刺激HER3的磷酸化和到Akt(细胞生存和增殖所需的一个关键的信号分子)的信号传递。在对EGFR抑制剂耐药的细胞中应用Met抑制剂,无论是体外还是体内实验,都促进了肿瘤细胞的凋亡、肿瘤生长速度的减缓以及显著的坏死。
Met and EGFR inhibitors combined together, cooperatively abrogate ErbB3 signaling activation (49). An alternative mechanism in this context is the Src-induced EGFR phosphorylation (52).
联合应用Met和EGFR抑制剂,能够协同地解除ErbB3的信号激活。另外一种机制是能阻止Src诱导的EGFR磷酸化。
Preclinical data also support that Met cross-talks and cooperates with other members of the EGF receptor family, including HER2, to enhance cell invasion and this lead to the possibility to explore therapeutic activity of dual Met and HER2 therapies (55,56).
临床前数据也支持Met与EGF家族其他成员的交互和协同作用,包括HER2,能够增强细胞的侵袭功能,这引起了对同时作用于Met和HER2的靶向治疗的探索。
Stimulation with both HGF and EGF enhances downstream activation of several signaling pathways including Akt, Erk and STAT3 in a way that Met inhibitors abolished their baseline phosphorylation (57,58).
HGF和EGF的共同刺激增强了下游多种信号通路的激活,其中包括Akt、Erk和STAT3,在这种情况下,Met抑制剂能够抑制它们的基础磷酸化。
The already mentioned interaction between decorin and LRIG1 proteins, promotes ligand-independent receptor downregulation and degradation of EGFR family members. Decorin binds to the EGFR family, inducing receptor dimerization, internalization and eventual lysosomal degradation, whereas LRIG1 and EGFR associated via their extracellular domains, allow enhanced EGFR phosphorylation. Thus, Met promotes resistance to VEGFR and EGFR inhibitors (59,60).
之前已经提到过胶饰蛋白多糖和LRIG1蛋白能够促进独立于配体的Met受体下调和EGFR族的降解。胶饰蛋白多糖绑定到EGFR家族,诱导了受体的二聚化、内在化和溶酶体降解。尽管LRIG1与EGFR结合与它们的胞外域,也能增强EGFR的泛素化。因此,Met促进了对VEGFR和EGFR抑制剂的耐药性。
Cross-talk between Met and KRAS signaling has also been described both in preclinical and clinical findings (61,62). Met activates RAS directly or via a protein-tyrosine phosphatase (63). Similarly, PI3K could be directly activated by Met or indirectly by RAS protein (30).
Met与KRAS的交互作用同样在临床前和临床研究中被发现。Met能够直接激活RAS,或者通过蛋白激酶磷酸化酶间接地激活。类似的,PI3K能够被Met直接激活或者通过RAS间接激活。
Moreover, Met directly binds to and sequesters the Fas receptor. This interaction prevents Fas self-aggregation and ligand binding, thus inhibiting Fas activation and apoptosis (64).
而且,Met能够直接绑定并且隔离Fas受体。这种相互作用阻止了Fas的自聚集和配体绑定,因此抑制了Fas的激活和细胞的凋亡。
Finally, preclinical studies exploring a combination of anti-Met therapeutic agents with mTOR inhibitors have also demonstrated an increased growth suppression, compared to mTOR inhibitors alone (62).
最后,临床前研究探索了联合抗Met治疗与mTor抑制剂的应用,相对于单独使用mTor抑制剂,这种方案同样展现了对肿瘤生长抑制效果的增加,
Met plays also a functional role in signaling pathways mediated by other membrane proteins. Integrin-dependent signaling could trigger ligand-independent Met phosphorylation following cellular adhesion, and Met and integrins might have independent yet synergistic roles in cell invasion. Plexins, single-pass transmembrane receptors for semaphorins, acts cooperatively with Met for cell adhesion and migration (45).
Met在由其他膜蛋白介导的信号通路中扮演了功能性角色。整合素依赖的信号能够在细胞粘附之后促发独立于配体的Met磷酸化,在细胞侵袭过程中,Met和整合素具有相互独立且协同增效的作用。Plexins,一种单次跨膜信号素受体,与Met协同作用于细胞的粘附和迁移
MET and NSCLC
MET与NSCLC
Met receptor is overexpressed in both Small Cell Lung Cancer (SCLC) and NSCLC, mainly in non-squamous histotype (65-67).
Met受体能在小细胞肺癌(SCLC)和非小细胞肺癌(NSCLC)中有过表达,在NSLCL中主要是非鳞癌。
Recent tumor microarray expression analysis demonstrated a 72% Met expression in human lung cancer tissue and 40% Met receptor over-expression; such values are higher than in breast (16%) and ovarian cancer (31%), but lower than in renal (70%) and colorectal cancers (CRC; 78%) (67). Phospho-Met expression is found to be at the highest levels in lung cancer (73%), followed by ovarian (33%), breast (23%), and renal (18%) cancer (67).
最近的肿瘤基因芯片表达分析表明,在72%的人类肺癌组织存在Met表达,有40%具有Met受体的过表达,该比例高于乳腺癌(16%)和卵巢癌(31%),低于肾癌(70%)和结直肠癌(78%)。磷酸化Met表达水平在肺癌中最高(73%),之后依次是卵巢癌(33%)、乳腺癌(23%)和肾癌(18%)。
Met gene amplification can guide the dependency of cell survival and proliferation upon the Met signaling, even in lung cancer cell lines. Blocking Met causes significant growth inhibition, G1-S arrest and apoptosis in cell lines harboring Met gene amplification. When Met is not amplifed, its levels of activation are low and cells are unable to grow (68)
在肺癌细胞系中,Met基因的扩展能够指导依赖于Met信号的细胞生存和增殖。在具有Met扩增的细胞系中,阻断Met能够导致显著的生长抑制, G1到S期的阻断和细胞的凋亡。当Met失去扩增时,其激活水平较低,细胞也无法生长。
Different studies have reported primary Met amplification to be in the wide range of 2% to 21%, in NSCLC lung adenocarcinomas, particularly in TKI-naive cohorts (69-72).
在肺腺癌中,特别是未接受过TKI的治疗的患者,不同的研究报道了原发的Met扩增的概率范围从2%到21%。
In lung cancer, Met receptor mutations were mainly found clustered in the non-tyrosine kinase domain, in the juxtamembrane (JM) domain and in the sema domain (67). These mutations are oncogenic activating variants, that result in a deletion in the juxtamembrane domain with enhanced oncogenic signaling, tumorigenicity, cell motility, and migration (27,73). Met kinase domain mutations have been found to be somatically selected in the metastatic tissues, compared with the primary solid cancers (74).
在肺癌中,Met受体突变主要聚集于非酪氨酸激酶域、近膜域以及sema域。这些突变是致癌激活的变种,这导致了近膜域的缺失从而增强了致癌信号传导、致瘤行、细胞移动以及迁移。相比原发病灶,Met激酶域的突变被发现有选择地存在于转移的组织中。.
Literature data are quite discordant on the prognostic value of Met over-expression, amplification and mutation.
数据表明,Met的过表达、扩增和突变对预后值来说存在不一致的影响。
The overexpression of circulating Met in patients with NSCLC has been strongly associated with early tumor recurrence and patients with adenocarcinoma and Met mplification have also demonstrated a trend for poor rognosis (69,75,76).
对于NSCLC患者来说,外周循环的Met过表达与早期肿瘤复发存在密切联系,肺腺癌患者和Met突变同样表现为较差的预后。
Concerning the correlation between Met FISH status and clinical characteristics, only Okuda and colleagues demonstrated an association with male gender and smoking status, showing also a relationship with high Met gene copy number (77). In the same trial, both FISH positive and gene amplified cases had a worse prognosis, although the difference was not statistically significant and among the Met FISH-positive NSCLCs, patients with gene amplification showed not significantly worse OS compared to those with high polysomy.
考虑到Met FISH状态与临床特征的联系,Okuda和同事发现,男性和吸烟状况与高Met拷贝数存在关联。在同一个临床试验中,同时具有FISH阳性和基因扩增的病例有更差的预后,虽然并未表现出统计学上的显著差异。在Met阳性的NSCLC患者中,相对具有多染色体型的患者,具有基因扩增的患者并未表现出显著降低的OS。
All FISH-positive cases had squamous histology, adenocarcinoma had Met amplification: high Met gene copy number tended to have shorter OS and PFS than those with low Met gene copy number, being this difference statistically significant only in the squamous histotype.
所有FISH阳性病例其组织学类型都是鳞癌,而腺癌具有Met扩增:高的Met基因拷贝数倾向于具有更短的OS和PFS,相比具有低拷贝数的患者;而这种差异只有在鳞癌中才表现得明显。
Moreover, at multivariate analysis done on squamous histology, increased Met gene copy number and Met amplification were confirmed to be independent poor prognostic factors.
进一步的,对鳞癌的多元分析显示,增加的Met基因拷贝数和Met扩增被确定为独立的较差的预后因子。
No significant difference in prognosis was found in atients having adenocarcinoma regardless Met FISH status in the korean study. In contrast, Beau-Faller and colleagues found a tendency toward shorter event-free survival in adenocarcinoma patients with increased Met gene copy umber, whereas Kanteti and colleagues demonstrated that the high Met gene copy number in adenocarcinoma was associated with a trend of better prognosis (69). However,
the above mentioned study has some critical methodology aspects as it was conducted on a small sample size and PCR was used as test and not FISH, done on DNA samples extracted from formalin-fixed paraffin-embedded (FFPE) archival tumor tissues (70).
来自韩国的研究显示,Met FISH的不同状态在肺腺癌患者中对预后的影响没有差异。与此相反的是,Beau-Faller与同事发现在具有增多的Met基因拷贝数的腺癌患者趋向于拥有较短的无事件生存期,但Kanteti与同事又发现在腺癌患者中,高Met基因拷贝数与更加的预后相关,但此研究存在一些方法学上的缺陷,一是样本太少,二是采用的PCR而不是FISH,来分析来自于存档肿瘤组织的福尔马林固定石蜡包埋样本中的DNA。
Capuzzo and colleagues found no patient with EGFR mutation was Met FISH positive, but increased Met gene copy number significantly correlated with EGFR FISH positive status (78).
Capuzzo与同事发现没有EGFR突变的患者同时具有Met FISH阳性,但Met基因拷贝数的增加与EGFR FISH阳性状态显著相关。
Acquired Met amplification has also been linked to approximately 22% of non-T790M mediated secondary gefitinib resistance in NSCLC patients, although it can also occur concurrently but independently (52,53,78-80).
获得性Met扩增存在于大约22%的非T790M介导的对吉非替尼耐药的NSCLC患者中,虽然Met与T790M能同时存在但它们彼此是独立的。
Using in vitro cell line models, the Met gene amplification in gefitinib-resistant cell clones was identified (53).
使用体外细胞系模型,Met基因扩增被发现存在于在对吉非替尼耐药细胞克隆中。
Rho and colleagues tried to demonstrate that Met activation, rather than gene amplification, is sufficient to promote EGFR resistance, but the activation appear to be secondary to increase passage numbers rather than to EGFR-Tki exposure (81).
Rho和同事尝试展示Met的激活状态而不是基因的扩增,是促进EGFR耐药的原因,但发现激活为传代数增加的次要表现,而不是对EGFR-TKi的暴露。
More recently, two prospective analyses have investigated the mechanism of EGFR-Tki resistance through the tissue rebiopsy: high Met gene copy number was found in 11% and 5% of the tissue samples, respectively (82,83).
最近,两个前瞻分析通过组织再活检来研究EGFR-TKI的耐药机制:高的Met基因拷贝数分别在11%与5%的组织样本中被发现。
FIN |
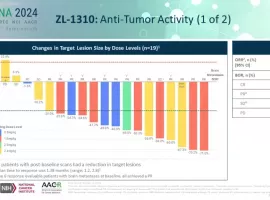 小细胞肺癌新希望?标准治疗耐药后OR
作者:Keenman
如果以药物规模化获批进入临床的时间来划分的话,进入21世纪以来,肺癌
小细胞肺癌新希望?标准治疗耐药后OR
作者:Keenman
如果以药物规模化获批进入临床的时间来划分的话,进入21世纪以来,肺癌
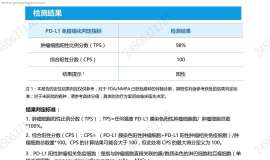
 母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下
母亲的基因检测和PD-L1报告出来了,
看了基因检测报告,似乎没有合适的靶向药,母亲确诊肺腺癌晚期,求助大家帮忙分析一下
 既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变
既要,又要,还要,什么药品可以治疗
作者:seacat
EGFR突变可以分为常见突变和罕见突变。常见突变就是EGFR外显子19del突变
 异常凝血酶升高是复发吗?
23年12月底确诊。24年1和2月做了介入,3月份切除,5月和6月做了预防性介入。7月到11月
异常凝血酶升高是复发吗?
23年12月底确诊。24年1和2月做了介入,3月份切除,5月和6月做了预防性介入。7月到11月
 晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半
晚期肺癌经过4次换药,如今母亲已经4
作者:pears
“万事开头难”这句话在母亲的抗癌路上体现得淋漓尽致,最初确诊肺癌后半





 显身卡
显身卡